Spatialproteomics: an interoperable toolbox for analyzing highly multiplexed fluorescence image data
May 3, 2025· ,,,,,,,,,,,,·
0 min read
,,,,,,,,,,,,·
0 min read
Matthias Meyer-Bender
Harald Vöhringer
Christina Schniederjohann
Sarah Koziel
Erin Chung
Ekaterina Popova
Alexander Brobeil
Lisa-Maria Held
Aamir Munir
Scverse Community
Sascha Dietrich
Peter-Martin Bruch
Wolfgang Huber
Abstract
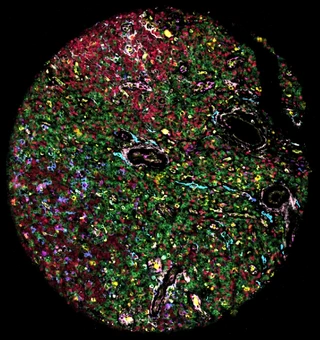
Highly multiplexed immunofluorescence imaging visualizes and quantifies protein levels at single-cell resolution in intact tissues at low cost and high scalability. Analysis of these data involves multiple steps with many method and parameter choices that must be adapted to the data and analytical objectives. There is an unmet need for a toolbox that offers flexible end-to-end coverage of the workflow.
Here we present spatialproteomics, a Python package that addresses these challenges. Spatialproteomics enables the processing and analysis of large imaging data, including steps such as segmentation, image processing, and cell type classification, while synchronizing shared coordinates across data modalities.
We demonstrate spatialproteomics on images of reactive lymph nodes and B cell Non-Hodgkin lymphomas (BNHL) from 132 patients. We showcase an end-to-end analysis from raw images to statistical characterization of how cell type composition and spatial distribution vary across indolent and aggressive lymphomas. Furthermore, we show how spatialproteomics can process Gigapixel whole-slide images.
Here we present spatialproteomics, a Python package that addresses these challenges. Spatialproteomics enables the processing and analysis of large imaging data, including steps such as segmentation, image processing, and cell type classification, while synchronizing shared coordinates across data modalities.
We demonstrate spatialproteomics on images of reactive lymph nodes and B cell Non-Hodgkin lymphomas (BNHL) from 132 patients. We showcase an end-to-end analysis from raw images to statistical characterization of how cell type composition and spatial distribution vary across indolent and aggressive lymphomas. Furthermore, we show how spatialproteomics can process Gigapixel whole-slide images.
Type
Publication
bioRxiv

Authors
Matthias Meyer-Bender
(he/him)
Bioinformatician
I am a PhD candidate at the European Molecular Biology Laboratory (EMBL) in the group of Wolfgang Huber. My current work focuses on developing open-source software for the processing and analysis of large-scale spatial omics datasets (spatialproteomics). I am passionate about applying computational methods, including machine learning and AI, to answer questions of biomedical relevance. Previously, I studied bioinformatics at the Technical University of Munich (TUM) and Ludwig Maximilian University of Munich (LMU). I also contribute to open-source projects such as scverse.
Authors
Authors
Authors
Authors
Authors
Authors
Authors
Authors
Authors
Authors
Authors
Authors